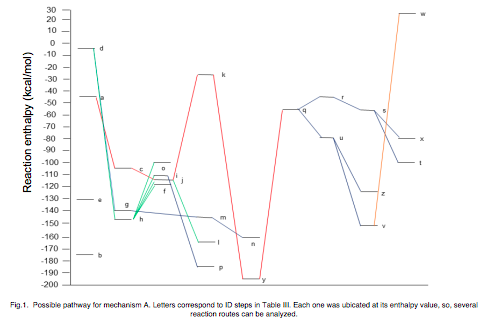
The photocatalytic degradation of ethylene over TiO2 has been widely studied, however, there are discrepancies between the degradation mechanisms proposed in experimental works. Some of them propose a degradation and mineralization mechanism trough ethoxide, acetaldehyde, acetic acid and finally carbon dioxide, whereas others did not find acetaldehyde or acetic acid, but formaldehyde and formic acid as intermediaries in the same process through the presence of the formyl radical HCOO on the catalyst surface. Through ab initio calculations it is possible to analyze the published experimental mechanisms in order to theoretically assess their feasibility and establish the possible reaction intermediaries and generated products. In this work, we used the Density Functional Theory based method DFT-RPBE/ 6-31G** in order to determine energy values to then estimate the enthalpy changes associated with each of the stages proposed for the ethylene degradation and mineralization processes, with which we elucidated the thermodynamically most probable mechanism, which explains differences between experimental work reports. We found that the most favorable route is through the formation of acetic acid, however, only one of the carbon atoms is converted to CO2, the other one is also converted to CO2 but from the formaldehyde degradation. These results agree with and explain those reported from experimental works. The method we used was validated by obtaining deviations shorter than 5% when comparing bond lengths, bond angles, dihedral angles, and vibrational frequencies calculated in this work versus experimental published values for most of the molecules involved.