The Physiome Project was initiated by the International Union of Physiological Sciences (IUPS) in 1997 to bring multiscale engineering modeling approaches to the physiological interpretation of the wealth of molecular data that was becoming available at that time [1]. The discipline of physiology, which with anatomy underpins medical practice, had lost its traditional central position in the biological sciences (at least from a funding perspective) to molecular biology, despite the very small impact molecular biology has had on the diagnosis and treatment of disease. While diseases and drugs certainly operate at the molecular level, the regulation of genetic transcription and, hence, the expression of proteins (the building blocks of life) are both highly dependent on environmental factors governed by the physical world in which molecular biology operates. Engineering—in particular, the rapidly growing field of bioengineering—is the discipline that has the integrative skills and tools to put the molecular pieces of Humpty Dumpty back together again.
In 2003, six years after the establishment of the Physiome Project by the IUPS, the U.S. Interagency Modeling and Analysis Group began funding physiome project grants. Four years later, in 2007, a consortium of European scientists and engineers published a roadmap, “Seeding the EuroPhysiome: A Roadmap to the Virtual Physiological Human” (VPH), that persuaded the European Commission to call for research proposals under Framework 7 on healthcare-related modeling. A VPH Institute (VPH-I) was subsequently established to provide continued global leadership for the VPH/Physiome Project with the goal of “[u]sing computational modeling of biological processes to integrate quantitative biological knowledge from molecular to cell, tissue, organ and whole body scales in order to understand physiological systems in terms of both their molecular components and their interaction with the environment and translate this understanding into clinical practice.” The VPH-I is also acting as a catalyst to bring together policymakers, science funding bodies, regulatory agencies, educational institutions, clinical organizations, and industry across the globe to maximize the benefit of VPH/physiome approaches for the healthcare industry and for the public good [2].
Mathematics is the language of quantitative science, and every scientific discipline should analyze its experimental data in the light of physical laws encapsulated in mathematical equations, but the challenge facing the nascent Physiome Project was the enormous complexity, including spatial and temporal scales, of molecular and physiological systems. Mathematical models of these processes quickly become difficult for anyone other than the author of a model to reproduce, let alone understand. The engineering solution to this problem is to build models based on standards and modularity. Data and modeling standards, with tools and model repositories based on these standards, ensure reproducibility and testability. And models must be built from modules that define the functional components of a system. Linking models of proteins to physiological scale models (i.e., cells, tissues, organs, and organ systems) also requires a hierarchy of semantically rich models so that an imported component module can be assembled automatically into the right place in the parent model.
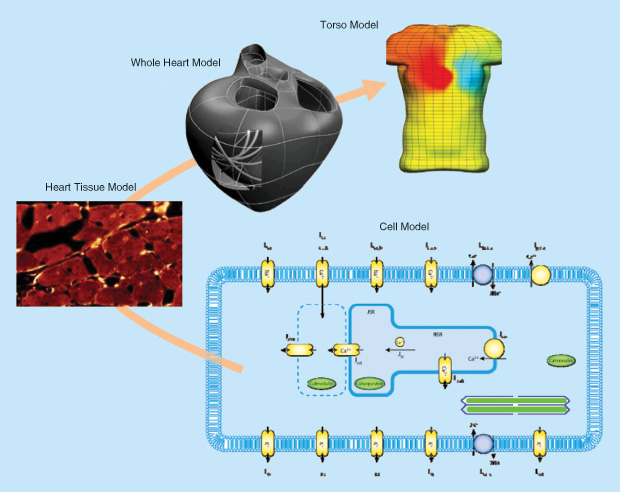
A typical multiscale model is shown in Figure 1 [3]. Four spatial scales are shown: the cell (cardiac myocyte), tissue (myocardium), organ (heart), and intact body (torso). The cell model shown here is, in fact, made up of many individual protein models. Heart disease and heart drugs operate at the protein level, but the diagnosis of disease is usually made at the level of the intact organ (e.g., the EKG for electrical function or ultrasound images for mechanical function). The challenge is to link models across these scales.
The initial focus for the Physiome Project was, therefore, the establishment of standards, based on modular components, that would help ensure reproducibility. The obvious choice of syntax for the model equations was MathML, the World Wide Web Consortium (W3C) standard for both content and presentation of mathematics. The need for semantically rich metadata was addressed through the Resource Description Framework (RDF), another W3C standard, using ontologies such as Chemical Entities of Biological Interest (ChEBI) for chemical identifiers, Gene Ontology (GO) for cell components, the CellType ontology, the Foundation Model of Anatomy (FMA), and the Ontology of Physics for Biology (OPB) for biophysical annotation. Based on these, two XML standards were developed: one called CellML for lumped parameter (not spatially varying) algebraic and ordinary differential equation (ODE) models such as the cell model shown in Figure 1 and another called FieldML for spatially varying (typically, finite element) models such as the tissue, organ, and torso models shown in Figure 1.
The CellML Standard
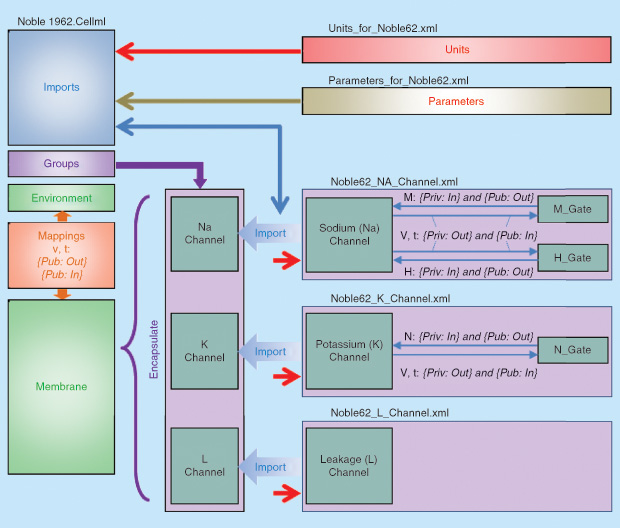
An example of the structure of a simple CellML model is shown in Figure 2. [A tutorial on creating and running this type of CellML model based on the freely available open source Physiome software OpenCOR is available online.] This cardiac electrophysiological model imports three ion channel models—a sodium channel (Noble62_Na_channel.xml), a potassium channel (Noble62_K_channel.xml), and a background leakage channel (Noble62_L_channel.xml)—that are grouped and encapsulated into a single higher-level membrane component in the parent model and combined with other components of the model. The units and parameters for the whole composite model are imported as separate modules and passed down as needed to the imported child modules. Note that CellML includes the base System International units and has facilities for constructing all derivative units [4].
There are about 600 publicly available CellML models covering a very wide spectrum of physiological processes (electrophysiology, excitation–contraction coupling, calcium dynamics, cell migration, cell mechanics, metabolic networks, signaling pathways, gene regulation, the cell cycle, circadian rhythms, etc.) on the Physiome Model Repository (PMR) at models.cellml. org. These models are all based on peer-reviewed published journal papers. Another 400 prepublication models are under development with password-protected workspaces designed for collaborative development of the models. The mathematics behind each of these CellML model exposures and workspaces can be rendered and automatically translated to various computer languages (C, Fortran, MATLAB, Python). Most of the CellML models available from the PMR have been curated to ensure that they produce results consistent with their reference publication. Some have also been annotated with RDF metadata to provide the biological and biophysical meaning of terms in the models.
The SED-ML Standard
Another standard called the Simulation Experiment Description Markup Language (SED-ML) has been developed to encapsulate the time duration and type of solver algorithm and its parameters together with model inputs and various outputs required by the simulation [5]. An example of this is shown in Figure 3 for solving the model shown in Figure 2. The SED-ML file also specifies the particular CellML model file that is being solved. A further XML standard that has been developed under the Physiome Project and implemented in OpenCOR is BioSignalML for time-varying signal data. Another modeling standard called the Systems Biology Markup Language (or SBML) designed for biochemical reactions, has been developed by the systems biology community.

Another initiative under the Physiome Project, called Web Lab, is the development of a resource that defines the protocols used for electrophysiological measurements of ion channel properties [6]. The process of testing a range of CellML models to see how well they capture the phenotype of a biological process—in this case, the current–voltage kinetics of membrane ion channels— is called functional curation and is an important development for the experimental validation of physiome models.
The Physiome Modeling Framework
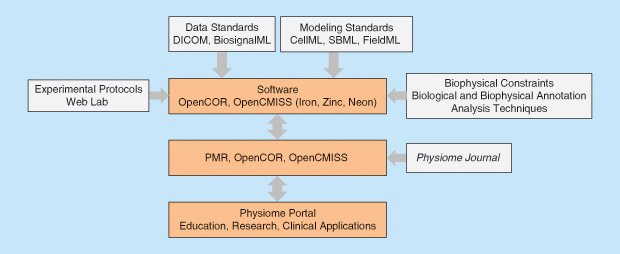
The overall framework for the VPH/Physiome Project is shown in Figure 4: at the top are the data and modeling standards; next is the open source software for creating, visualizing, and simulating both the ODE-based (CellML) and the partial differential equation (PDE)-based (FieldML) models using, respectively, OpenCOR and OpenCMISS; then comes the PMR; and, finally, there is a physiome portal (still under construction).
At present, the OpenCOR software used to author and curate CellML models and run simulations with them using SED-ML scripts checks that units are consistent throughout the equations and, when annotated with semantic RDF metadata, will ensure that the mathematical terms are consistently labeled with terms from the community defined ontologies (ChEBI, GO, CellType, FMA, OPB). A tool called Semgen is used to create composite RDF strings that combine the biological and biophysical annotations [7]. These features provide a powerful tool for biological modeling—nearly all of the 600 models in PMR required terms missing from the journal publications to be added or mistakes in the equations to be rectified to achieve a working model that produced output matching the graphs in the publications. The modularity built into the design of CellML is another powerful aspect that encourages good practice.
None of this, however, ensures that the CellML models are valid or robust. A validated model is one that has been successfully tested against a range of experimental data—and the Web Lab resource mentioned previously is designed to assist with validation. A robust model, on the other hand, is one that satisfies biophysical constraints, such as electrical charge conservation or the Gibb’s free energy changes demanded by the second law or thermodynamics. Robustness also means that when such a model is imported as a module into a higher level model, these biophysical properties are maintained.
A promising approach for ensuring robustness in modeling is provided by Bond graph theory [8]. The five types of physical driving force (called effort in the Bond graph literature) commonly encountered in CellML models are electrical potential, mechanical force, mechanical torque, hydrostatic pressure, and chemical potential. The basic units upon which these driving forces operate are, respectively, the coulomb (C), length (m), angle (radian), volume (m^3), and moles of solute (mol). The common currency here is energy in joules (J), and the driving force can, in each case, be written as energy per relevant unit on which the driving force acts. These are, respectively, J·C ^-1 (or volts), J·m ^-1 (or Newtons), J·rad ^-1 (or N·m·rad ^-1), J·m ^-3 (or Pascals), and J·mol ^-1.
In each case, there is a flux or rate of flow (called flow in the Bond graph literature) of the relevant unit: C·s ^-1, m·s ^-1, rad·s ^-1, m3·s ^-1, and mol·s ^-1. The product of the driving force (effort) and the flux (flow) is always power (J·s ^-1 or watts). The key notion in the Bond graph approach is energy transfer between different physical systems. A Bond graph is a collection of lines, each of which is associated with a driving force (effort) and flow and nodes that are either ideal effort sources (unaffected by the flow) or flow sources (unaffected by the effort). By separating energy generation, transmission, storage, and dissipation, Bond graphs enforce a clear distinction between model parameters that are associated with biophysical laws (such as thermodynamic constraints) and those that are associated with empirical relations that approximate material behavior [8]. The Bond graph framework is being implemented in OpenCOR as an optional template that modelers can use to construct (for example) thermodynamically robust models.
The combination of the CellML and SED-ML standards also makes it possible to construct useful modeling templates. For example, a template is being developed for use in OpenCOR that will help electrophysiology modelers implement ion channel kinetics with Markov state models by automatically constructing the transition probabilities from the state diagrams.
Future Developments
New Physiome Project initiatives, being developed both by the IUPS Physiome Committee and the VPH Institute, are 1) the establishment of a new journal, the Physiome Journal; 2) a new web portal, based on models from the Physiome Journal, that provides access to integrative whole body multiscale physiome models; and 3) a mapping from CellML models to electronic health records (EHRs) and biobanks. The IUPS Physiome Journal will solicit papers that describe models and associated data encoded in physiome standards and should complement journals that describe physiological experiments and the use of models to interpret these experiments. The prospect of citable articles that fully document a model for the benefit of others should provide an incentive for modelers to make the effort to ensure that their models are curated, annotated, and modular—because they will then be getting a second citable paper to complement the one submitted to the existing journals. At the moment, there is no incentive for most modelers to make that effort.
The second proposal for a web portal is being developed by the VPH Institute such that models published by the Physiome Journal would be automatically linked into the evolving wholebody physiological models (including whole-body circulation models and physiologically based pharmacokinetic and pharmacokinetic-pharmacodynamic models) available on the VPH portal. One role for the portal will be to identify areas where there are no models and to encourage new experimental and modeling efforts to fill those gaps.
The third proposal is designed to use the physiome models, with patient-specific parameters, as a quantitative statement of phenotype that can be incorporated into EHRs via the openEHR standard. Links are also being developed with biobanks to assist with the interpretation of longitudinal clinical data.
In summary, good practice requires biological models to be 1) reproducible; 2) consistent in their units; 3) modular; 4) semantically annotated; 5) accessible from free, open-source repositories; 6) usable with standards- based authoring and simulation software; 7) validated; and 8) biophysically robust. The Physiome Project has achieved 1) through 6) for ODE and algebraic models and is currently well on track to achieve the last two objectives over the next few years. The development of the FieldML standard for spatially dependent models is also progressing but is a much harder problem. By helping in the development of robust, reproducible, multiscale models, the Physiome Project should help put the biological sciences and medical diagnostics onto a more quantitative and biophysically rational footing, in line with all physical and engineering sciences.
Acknowledgment
am grateful to my colleague, Prof. Stig Omholt, for pointing out that, around 150 years ago, an editorial in an important electrical trade journal stated [9]: “We may remark that of late years the (experimental) facilities for obtaining definite ideas on the subject of electrical phenomena have greatly increased. Sciences generally become simplified as they advance, albeit in some branches they may attenuate into the abstruse. In electricity there is seldom any need of mathematical or other abstractions, and although the use of formulae may in some instances be a convenience, they may for all practical purposes be dispensed with.” Considering the role that mathematics (and Maxwell’s equations in particular) since then has played for technology development based on electrical phenomena— and on which our civilization is completely dependent— we all laugh at this. But what if we change the phrase “the subject of electrical phenomena” with “the subject of biological phenomena” and “electricity” with “biology”?
References
- P. J. Hunter and T. K. Borg, “Integration from proteins to organs: The Physiome Project,” Nature Rev. Mol. Cell Biol., vol. 4, no. 3, pp. 237–243, 2003.
- P. Hunter, T. Chapman, P. V. Coveney, B. de Bono, V. Diaz, J. Fenner, A. F. Frangi, P. Harris, R. Hose, P. Kohl, P. Lawford, K. McCormack, M. Mendes, S. Omholt, A. Quarteroni, N. Shublaq, J. Skår, K. Stroetmann, J. Tegner, S. R. Thomas, I. Tollis, I. Tsamardinos, J. H. G. M. van Beek, and M. Vicecont, “A vision and strategy for the virtual physiological human: 2012 update,” Interface Focus, vol. 3, p. 20130004, 2013.
- P. J. Hunter, A. J. Pullan, and B. H. Smaill, “Modeling total heart function,” Annu. Rev. Biomed. Eng., vol. 5, pp. 147–177, 2003.
- A. A. Cuellar, C. M. Lloyd, P. F. Nielsen, M. D. B. Halstead, D. P. Bullivant, D. P. Nickerson, and P. J. Hunter, “An overview of CellML 1.1, a biological model description language. SIMULATION,” Trans. Soc. Model. Sim. Int., vol. 79, no. 12, pp. 740–747, 2003.
- D. Waltemath, R. Adams, F. T. Bergmann, M. Hucka, F. Kolpakov, A. K. Miller, I. I. Moraru, D. Nickerson, S. Sahle, J. L. Snoep, and N. Le Novère, “Minimum information about a simulation experiment (MIASE),” PLoS Comput. Biol., vol. 7, no. 4, p. e1001122, 2011. doi:10.1371/journal.pcbi.1001122
- J. Cooper, M. Scharm, and G. R. Mirams, “The cardiac electrophysiology web lab.,” Biophys. J., vol. 110, pp. 292–300, 2016. doi:10.1016/ j.bpj.2015.12.012
- M. L. Neal, M. T. Cooling, L. P. Smith, C. T. Thompson, H. M. Sauro, B. E. Carlson, D. L. Cook, and H. H. Gennari, “A reappraisal of how to build modular, reusable models of biological systems.,” PLoS Comput. Biol., vol. 10, no. 10, p. e1003849, 2014. doi:10.1371/journal.pcbi.1003849
- P. J. Gawthrop and E. J. Crampin, “Energy-based analysis of biochemical cycles using bond graphs.,” Proc. R. Soc. A, vol. 470, 2014. doi:10.1098/rspa.2014.0459
- P. J. Nahin, Oliver Heaviside: The Life, Work, and Times of an Electrical Genius of the Victorian Age. London, UK: The Johns Hopkins University Press, 2002.