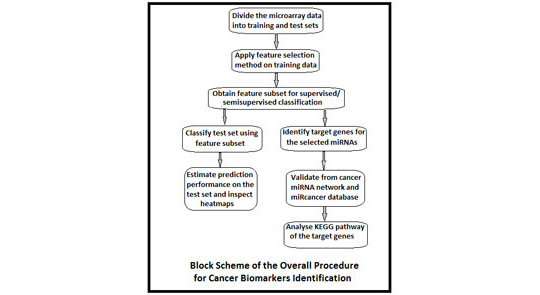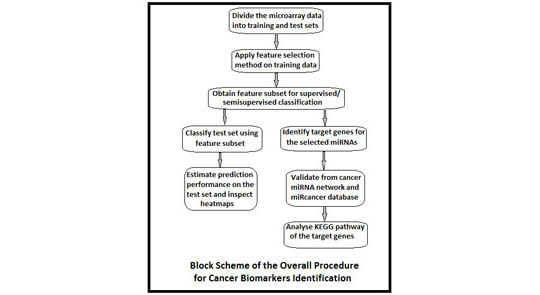
Microarrays have now gone from obscurity to being almost ubiquitous in biological research. At the same time, the statistical methodology for microarray analysis has progressed from simple visual assessments of results to novel algorithms for analyzing changes in expression profiles. In a micro-RNA (miRNA) or gene-expression profiling experiment, the expression levels of thousands of genes/miRNAs are simultaneously monitored to study the effects of certain treatments, diseases, and developmental stages on their expressions. Microarray-based gene expression profiling can be used to identify genes, whose expressions are changed in response to pathogens or other organisms by comparing gene expression in infected to that in uninfected cells or tissues. Recent studies have revealed that patterns of altered microarray expression profiles in cancer can serve as molecular biomarkers for tumor diagnosis, prognosis of disease-specific outcomes, and prediction of therapeutic responses. Microarray data sets containing expression profiles of a number of miRNAs or genes are used to identify biomarkers, which have dysregulation in normal and malignant tissues. However, small sample size remains a bottleneck to design successful classification methods. On the other hand, adequate number of microarray data that do not have clinical knowledge can be employed as additional source of information. In this paper, a combination of kernelized fuzzy rough set (KFRS) and semisupervised support vector machine (S³VM) is proposed for predicting cancer biomarkers from one miRNA and three gene expression data sets. Biomarkers are discovered employing three feature selection methods, including KFRS. The effectiveness of the proposed KFRS and S³VM combination on the microarray data sets is demonstrated, and the cancer biomarkers identified from miRNA data are reported. Furthermore, biological significance tests are conducted for miRNA cancer biomarkers.
View full article